PUDJIATMOKO, DVM., Ph.D
Veterinary Drug Assay Laboratory
(Balai Besar Pengujian Mutu dan Sertifikasi Obat hewan)
Gunung sindur, Bogor 16340
ABSTRACT
Molecular identification of 6 species of family Chlamydiaceae carried out by nested polymerase chain reaction (PCR) with species-specific primers derived from 16S rRNA gene was investigated. The 16SA and 16SB primers were used for amplification of 1,473 bp 16S rRNA gene fragments of 25 strains of Chlamydiaceae in the first PCR. A set of family-specific primer and 5 sets of species-specific primer were used for amplification of fragments of 16S rRNA gene in the nested PCR. The family-specific primers amplified all the 16S rRNA gene of Chlamvdophila spp. used. The speciesspecific primers were shown to be specific for each species. Multiplex PCR calTied out using 3 primers (2 sense primers and I antisense primer) in a single tube was applicable for detection of Chlamydophila felis, Chlamydophila caviae and Chlamydophila psittaci. C. psittaci-specific primers could be used for detection of Chlamydophila DNA extracted from 1 throat swab of human, 1 liver specimen of a parakeet and 2 fecal specimens of crows. C. felis specific primers could be used for detection of Chlamydophila DNA extracted from 4 throat swabs of humans and 2 conjunctival swabs of cats.
Key words: Chlamydiaceae, Chlamydophila, multiplex PCR
ABSTRAK
Identifikasi molekul 6 spesies famili Chlamydiaceae dengan cara nested polymerase chain reaction (PCR) telah dilakukan dengan menggunakan primer spesifik-spesies berasal dari gen 16S rRNA. Primer 16SA dan 16SB digunakan untuk mengamplifikasi 1.473 bp gen 16S rRNA dari 25 galur Chlamydiaceae pada PCR pertama. Satu set primer spesiflk-famili dan 5 set primer spesiflk-spesies digunakan untuk mengampliflkasi sebagian gen 16S rRNA pada nested PCR. Plimer spesiflk-famili dapat mengamplifikasi semua gen 16S rRNA dari Chlamlydophila spp. yang digunakan. Primer spesifik spesies menunjukkan spesifik untuk setiap spesies. PCR multiplek menggunakan 3 primer (2 primer sense clan I primer antisense) dalam satu tabung dapat diaplikasikan untuk mendeteksi Chlamydophila felis, Chlamydophila caviae dan Chlamydophila psittaci. Primer spesifik-C. psittaci dapat digunakan untuk mendeteksi DNA Chlamydophila yang diekstraksi clari I swab tenggorokan manusia, I spesimen hati burung pal'kit, clan 2 spesimen feses gagak. Primer spesifik-C. felis dapat digunakan untuk mendeteksi DNA Chlamydophila yang diekstraksi dari 4 swab tenggorokan manusia dan 2 swab selaput kelopak mata kucing.
Kata kunci : Chlamydiaceae, Chlamydophila, PCR multipleks
INTRODUCTION
Chlamydophila causes a wide variety of diseases in animals and humans (Fukushiand Hirai, 1992; Fukushi and Hirai, 1993a; Grayston et al., 1989; Moulder et al., 1984). The genetic diversity of Chlamydophila is well recognized by chromosomal DNA finger printings (Fukushi and Hirai, 1989), restriction fragment length polymorphism (RFLP) of rRNA gene loci (Fukushi and Hirai, 1993b), RFLP of ompA and groEL genes (Fukushi and Hirai, 1994), and random amplification of polymorphic DNA (RAPD) analysis (Pudjiatmoko et at., 1996). Chlamydophila classified into several genetic groups based on the ompA gene sequence (Kaltenboeck et at., 1993). The previous phylogenetic analysis of the genus Chlamydophila based on 16S rRNA gene sequences showed 6 genetic groups (Pudjiatmoko et aI., 1997). In 1999 the genus Chlamydophila was classified into 6 species: Chlamydophila psittaci, C. felis, C. caviae, C. abortus, C. pecorum and C. pneumoniae (Everett et aI., 1999). A practical method for genotyping Chlamydophila is not available yet.
Typing of Chlamydophila isolates has traditionally been done by immunological methods including fluorescent antibody and enzyme immunoassays (Fukushi and Hirai, 1989; Stamm et at., 1988; Storz and Krauss, 1985; Thomas et aI, 1990). Recently, m9lecular methods using gene amplification, LCR, and DNA sequencing have been employed for identification of isolates (Campbell et aI., 1993; Domeika et aI., 1994; Gaydos et aI., 1992; Holland et aI., 1990; Kaltenboeck et aI., 1991; Kaltenboeck et al., 1993; Kaltenboeck and Storz, 1992; Mahony et aI., 1993). This report describes a simple method of genetic typing of Chlamydophila by PCR using species-specific primer sets. Multiplex PCR carried out using 3 primers (2 sense primers and 1 antisense primer) in a single tube was applicable for detection of C. felis, C. caviae and C. psittaci. Furthermore, this method was applied for PCR and genetic typing of Chlamydophila spp. from clinical samples of human and animal.
MATERIALS AND METHODS
Microorganisms
Chlamydophila strains used in this study included 5 strains (Frt-Hu/Cal 10, Hu/Borg, Prk/6BCT, Prk/ Daruma, Prt/GCPI) of C. psittaci, 1 strain (Gp/Ie) of C. caviae, 7 strains (Fe/Pnl, Fe/ 145, Fe/Cello, Fe/B 166, Fe/C 164, Fe/C429, Fe/C454) of C. felis, 5 strains (Bo/ESST, Bo/Maeda, Bo/Shizuoka, Koala (type 2), Ov/IPA) of C. pecorum, 1 strain (TW/83T) of C. pneumoniae and 6 strains (A/Har-l T, B/TW-5/0T, C/ TW-3/0T, DfUW-3/Cx, EfUW-5/Cx, L2/434/Bu) of C. trachomatis (Pudjiatmoko et aI., 1997). To confirm the specificity of PCR, the following agents were tested: Staphylococcus aureus, Streptococcus pneumoniae (Gifu 8766 strain), Bordetella bronchoseptica (Gifu 1127 strain), Mycoplasma pneumoniae, Escherichia coli (C600 strain), Haemophilus influenzae (Gifu 3191 strain), Klebsiella pneumoniae (Gifu 2929 strain), Leginella pneumophila (SL94-1 strain), Leginella pneumophila (SL94-2 strain), Coxiella burnetii (Nine Mile ATCC YR 615 strain), Orientia tsutsugamushi (Gilliam Strain), Orientia tsutsugamushi (Karp strain) and Orientia tsutsugamushi (Kato strain). These strains were kindly provided by Dr. T. Ezaki of Gifu University, Japan and Dr. A. Tamura of Niigata College of Pharmacy, Japan.
Clinical samples
One hundred and forty throat swabs were collected from junior high school students with respiratory diseases. Eleven conjunctival swabs were collected from cats suffering conjunctivitis at the animal hospital of Gifu University and Tokyo Metropolitan Institute. Thirty-three fecal samples from crows and ten liver tissue samples from parakeets with systemic infections were also tested.
Preparation of chlamydial DNA
Procedures for preparation of Chlamydophila DNA have been described previously (Fukushi and Hirai, 1989). Extraction of chlamydial DNA from throat swabs, sera and fecal samples was performed according to the protocol of Higuchi (Higuchi, 1989). Briefly, all the samples were suspended in 1 ml of PBS in a 1.5 ml Eppendorf micro centrifuge tube, and centrifuged at 13,000 x g for 20 sec. The pellet was resuspended in 1 ml of washing buffer (10 mM TrisHCI (pH 8.3), 50 mM KCI, 1.5 mM MgC12), and centrifuged at 13,000 x g for 20 sec. The pellet was resuspended in 1 ml lysis buffer (0.32 M sucrose, 10 mM tris-HCI (pH 7.5), 1 % Triton X-lOO), and centrifuged at 13,000 x g for 20 sec. DNA was released from the pellet by adding PCR buffer with nonionic detergents (50 mM KCI, 10 mM Tris-HCI (pH 8.3), 2.5 mM MgCI2, 0.1 mg/mi gelatin, 0.45 % NP40 and 0.45 % Tween 20), and proteinase K (120 pg/ml) to the washed pellet. The mixture was incubated for 1 h at 55° C, boiled for 10 min, and then chilled on ice.
Nucleotide primers
For amplifying chlaniydial 16S rRNA gene, a pair of primer 16SA-16SB, was constructed from the 16S rRNA sequences of the family Chlamydiaceae (Pudjiatmoko et at., 1997). Internal primers were prepared from the 16S rRNA gene from the C. felis, C. psittaci, C. pecorum, C. pneumoniae and C. trachomatis for the nested PCR. The primers were designated as ChFeA-ChFeB, ChPsA-ChPsB, ChPeA-ChPeB, ChPnA-ChPnB, and ChTrA-ChTrB primers for the C. felis, C. psittaci, C. pecorum, C. pneumoniae and C. trachomatis, respectively (Table 1). The combination of ChFeA-ChPsA-ChPsB primers was used in a one-step reaction carried out in a single tube for genotyping of C. caviae, C. felis and C. psittaci. The length of the DNA fragments generated in the PCRs are shown in Table 2.
PCR amplification of clinical samples
Chlamydopila 16S rRNA genes were amplified by using a pair of 16SA and 16SB primers in the first PCR. The PCR was performed with 5 ul of throat swab, fecal, cell culture samples or chromosomal DNA of Chlamydopila extracts in a total volume of 50 ul. Five ul of the first PCR product was submitted to the nested PCR using a pair of family- and species-specific DNA primer. The final reaction mixture contained 40 pmol of each primer, 10 mM Tris-HCl (pH 8.3), 50 mM KC1, 1.0-2.0 mM MgCI2, 200 pM each of dATP, dCTP, dGTP, and dTTP, 0.01 % gelatin, and 2.5 units of Taq polymerase (Takara Shuzo Co., Ltd., Kyoto, Japan) in 50 ul. Concentration of MgCl2 was 1.0 mM for ChPsA-ChPsB, ChFeA-CbPsB and ChTrA-ChTrB primers, 1.5 mM for 16SA-16SB, ChA-ChB, and ChPnA-ChPnB primers, and 2.0 mM for ChPeAChPeB primers. The components and procedure for the multiplex PCR were described above except 2 sense primers for C. felis (ChFeA) and C. psittaci (ChPsA) and an antisense primer for C. psittaci (ChPsB) was used in the method. The thermal cycle amplification program for the first PCR was as follows: initial melting at 94°C for 3 min and then 35 cycles with denaturation at 94°C for 1 min, annealing at 55°C for 1 min and extension at 72°C for 2 min and the last extension for 7 min follow by holding at 4°C. The program for nested PCR was as follows: initial melting at 94°C for 3 min and then 35 cycles with denaturation at 94°C for 30 see, annealing at 55°C for 30 sec and extension at 72°C for 2 min and the last extension for 7 min follow by holding at 4 C. Five ul PCR products were electrophoresed in 2 % agarose gel. DNA was stained with ethidium bromide and visualized by UV light.
Table 2. Specificity of peR test of 5 species-spesific primer sets of Chlamydophila
Isolation of Chlamydophila
Clinical specimens were ground to make a 10 % emulsion in SPG containing 500 ug/ml streptomycin and Kanamycin. The mixtures were clarified by centrifugation. The supernatant fluid was inoculated into yolk sac of specific pathogen-free embryonated chicken eggs that had been incubated at 37°C for 67 days. The inoculated eggs were incubated at 37°C for 3 - 7 days, and yolk sac smears were microscopically examined for viable Chlamydiae after staining by the Gimenez method (Schachter, 1988).
RESULTS
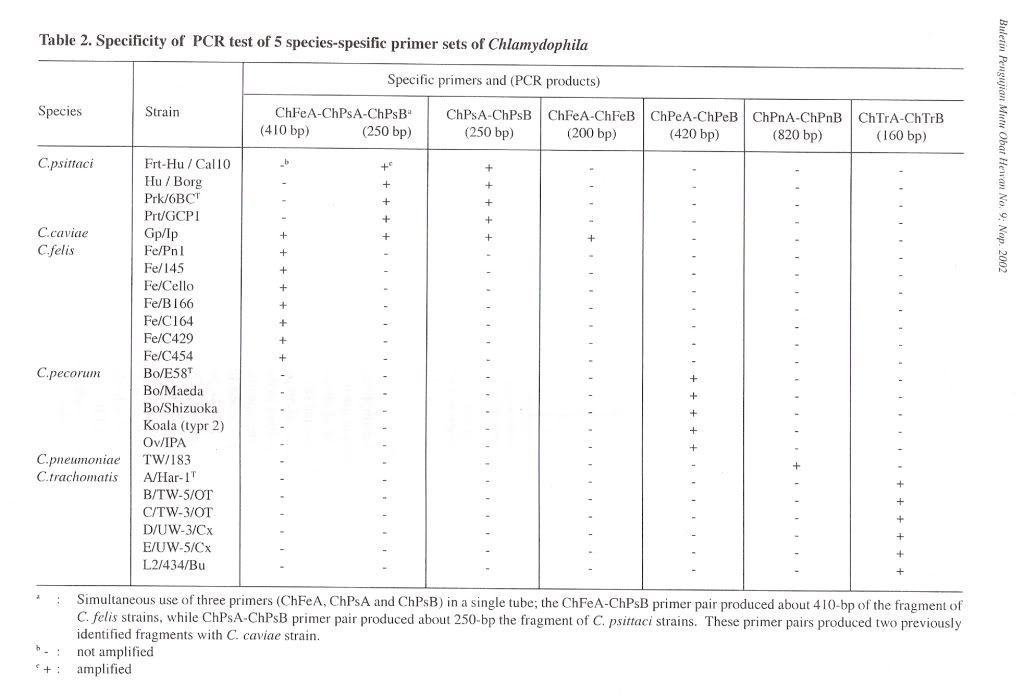
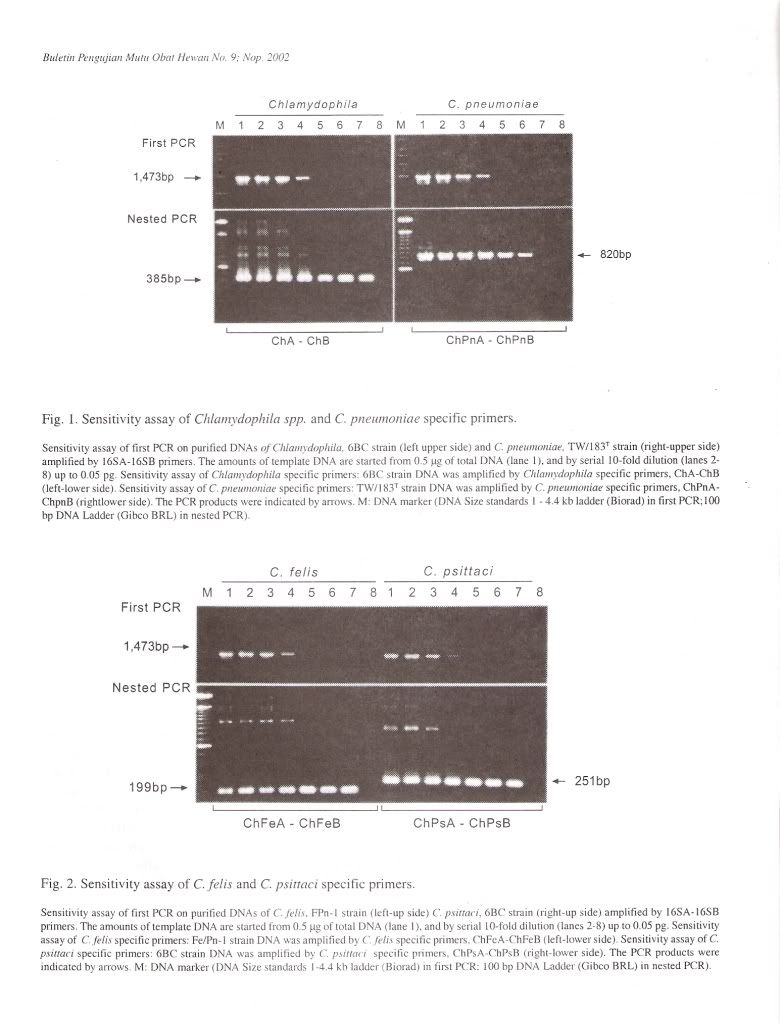
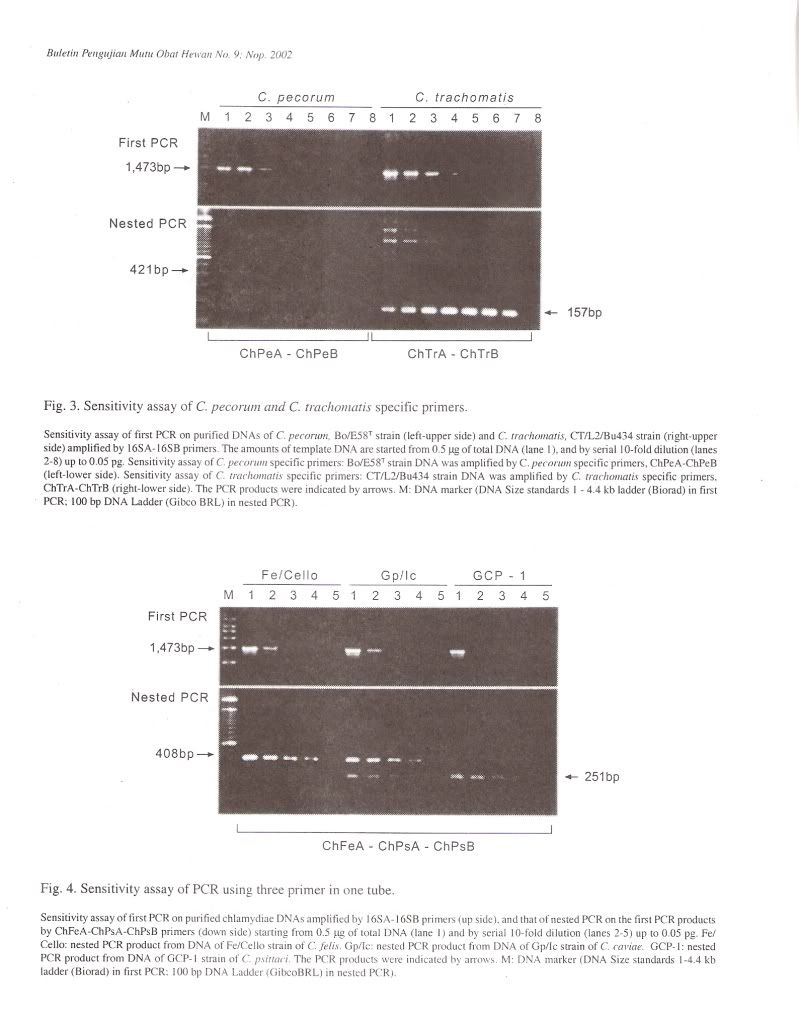
Specificity of PCR amplificationTo evaluate the specificity of our primer sets, 25 Chlamydophila and 2 Gram-negative and 7 Grampositive bacterial and 4 Rickettsial strains were tested in this study. In the first PCR, the 16S rRNA gene fragment of all Chlamydophila strains was amplified by the family-specific primer set of 16SA-16SB, and the size of PCR product was about 1470 bp (Figure 1). In the second PCR, the 16S rRNA gene fragment of all Chlamydophila DNA was examined by amplification with the family specific-primer set, ChA-ChB. The size of PCR product was about 390 bp (Figure 1). A DNA product of approximately 820 bp in size was observed with C. pneumoniae DNA with the primer set ChPnA-ChPnB. DNA products with approximately 200 bp and 250 bp were observed with the C. feils and C. psittaci DNA with the primer sets of ChFeA-ChFeB and ChPsA-ChPsB, respectively (Figure 2). DNA products with approximately 420 bp and 160 bp were observed with C. pecorum and C. trachomatis DNA with the primer sets of ChPeA-ChPeB and ChTrA-ChTrB, respectively (Figure 3). The 16S rRNA gene fragments of C. caviae were amplified by primer sets of ChFeAChFeB and ChPsA-ChPsB, or ChFeA-ChPsB. The 16S rRNA gene fragment of C. felis was also amplified by primer set of ChFeA-ChPsB. Thus the species specific sets were specific for each species except guinea pig (Table 2). All 2 Gram-positive and 7 Gram-negative bacterial and 4 Rickettsial strains tested yielded no PCR product with all primer pairs used in both the first and second PCR.
Multiplex PCR
To simplify PCR reaction, co-amplification was carried out by simultaneous use of three primers (ChFeA, ChPsA and ChPsB) ina single tube. In this reaction, the ChFeA-ChPsB primer pair produced about 410 bp of the fragment of , C. felis strains, while ChPsA-ChPsB primer pair produced about 250 bp fragment of C. psittaci strains (Figure 4). These primer pairs produced two previously identified fragments with C. caviae strain. However, Chlamydophila strains from other genetic groups did not produce any PCR products.
Sensitivity of PCR amplification
The sensitivity of the PCR method developed in the present work was evaluated by testing serial dilution of purified DNA from 25 Chlamydophila strains. In the first PCR, PCR signals from 0.5 pg to 50 ug of template DNA could be detected in 50 ul aliquots of reaction. When the first PCR products were submitted to nested PCR, detectable levels were from 5 fg to 50 ng of the original template Chlamydophila DNA (Figure 4).
Direct detection of Chlamydophila from clinical specimens
After the specificity and sensitivity of the PCR method were determined, the evaluation of direct detection and genotyping of Chlamydophila from clinical samples was done simultaneously. Chlamydophila organisms were detected by multiplex PCR in 1 liver specimen from 10 parakeets, 2 conjunctival swabs from 11 cats, 2 fecal specimens from 33 crows, and 5 throat swabs from 140 humans. PCR results in parakeets and cats were confirmed by isolation. Isolation was not attempted with crow and human specimens. The species o~ these ten positive PCR amplified DNAs were determined to be a C. psittaci from parakeet, two C. psittaci from crows, a C. psittaci from human, a C. felis from cat, a C. felis from parakeet, and four C. felis from humans. The genotype of isolates from parakeets and cats confirmed the PCR typing results.
DISCUSSION
The PCR using primers targeted to the 16S rRNA gene were developed for direct identification of Chlamydophila spp. in the present study. The specificity of the amplifications was confirmed by sequencing of DNA from PCR amplified products and isolates from clinical samples. Using a primer set for the family Chlamydiaceae in combination with five primer sets for the existing species C. caviae, C. felis, C. psittaci, C. pecorum, C. pneumoniae, and C. trachomatis, each designated chlamydial species was able to be differentiated. The method was applied for detection and direct typing of Chlamydophila for clinical samples from humans and animals. The results of the study demonstrated that this method may be useful for laboratory diagnosis.
The sensitivity of our method that detect 1 to 50 IFU or 5 to 50 fg is equivalent to other method that use the MOMP gene (Campbell et al., 1992; Valassina et al., 1995), l6S rRNA gene (Gaydos et al., 1992) and plasmid (Valassina et al., 1995). The PCR with species-specific primer pairs could identified species of Chlamydophila. However, no amplification was observed with other bacteria in both of the PCR. The results indicated that the PCR methods revealed specific to detect family Chlamydiaceae and identify genetic groups of Chlamydophila.
Currently 3 genetic groups are recognized for C. Caviae, C. felis and C. psittaci (Pudjiatmoko et al., 1997; Everett et aI., 1999). Differentiation ofthese 3 genetic groups requires 6 primers (3 sense and 3 antisense primers) using 3 sets of tubes. In method of this study, PCR was carried out using three primers (two sense primers and an antisense primer) in a single tube for both detect and identify species C. caviae, C. felis, and C. psittaci simultaneously. This multiplex PCR method uses less reagent, time and labor. The multiplex PCR technique appears to be suitable method for a rapid detection and genotyping of Chlamydophila isolates.
No case of feline chlamydiosis in human has been reported in Japan. It is also rare in other countries. A human case of feline pneumonitis was reported in 1969 (Schachter et al., 1969). Our study found 4 out of 140 high school children have feline pneumonitis. This result indicate that transmission of feline pneumonitis from cats to human is not rare and infection is often subclinical.
ACKNOWLEDGMENT
This study was supported by grants-in-aid for Scientific Research from the Japanese Ministry of Education, Science, Sports and Culture.
REFERENCES
Campbell, L.A., M.P. Melgosa, D.J. Hamilton and C.-C. Kuo. 1992. Detection of Chlamydia pneumoniae by polymerase chain reaction. J. Clin. Microbiol. 30: 434439.
Campbell, L.A., D.L. Patton, D.E. Moore, A.L.
Cappuccio, B.A. Mueller and S.-P. Wang. 1993. Detection of Chlamydia trachomatis deoxyribonucleic acid in women with tubal infertility. Fertil. Steril. 59: 45-50.
Domeika, M., A. Ganusauskas, M. Bassiri, G.
Froman and P.-A. Mardh. 1994. Comparison of polymerase chain reaction, direct immunofluorescence, cell culture and enzyme immunoassay for detection of Chlamydia psittaci in bull semen. Vet. Microbiol. 42: 273-280.
Everett, K.D.E., R.M. Bush and A.A. Andersen. 1999. Emended description of the order Chlamydiales, proposal of Parachlamydiaceae fain. novo and Simkaniaceae fain. nov., each containing one monotypic genus, revised taxonomy of the family Chlamydiaceae, including a new genus and five new species, and standards for the identification of organisms. Int. I. Syst. Bacteriol. 49: 415-440.
Fukushi, H. and K. Hirai. 1989. Genetic diversity of avian and mammalian Chlamydia psittaci strains and relation to host origin. J. Bacteriol. 171: 2850-2855.
Fukushi, H. and K. Hirai. 1992. Proposal of Chlamydia pecorum sp. novo for Chlamydia strains derived from ruminants. Int. J. System. Bacteriol. 42: 306-308.
Fukushi, H. and K. Hirai. 1993a. Chlamydia pecorum - The forth species of genus Chlamydia Microbiol. Immunol. 37: 5 15-522.
Fukushi, H. and K. Hirai. 1993b. Restriction fragment length polymorphisms of rRNA as genetic markers to differentiate Chlamydia spp. Int. J. Syst. Bacteriol. 43: 613-617.
Fukushi, H. and K. Hirai. 1994. Heterogeneity and homogeneity of ompA (the major outer membrane protein) and GroEL-homolog genes of avian and mammalian Chlamydia psittaci by PCR-based RFLP analysis. In: Orfila, J., G.I. Byrne, M.A. Cherne sky, J.T. Grayston, R.B. Jones, G.L. Ridgway, P. Saikku, J. Schachter, W.E. Stamm and R.S. Stephens (eds.). Eighth International Symposium on Human Chlamydial Infections. pp. 589-592.Societa Editrice Esculapio, Bologna.
Gaydos, e.A., T.e. Quinn and J.J. Eiden. 1992 ..
Identification of Chlamydia pneumoniae by DNA amplification of the 16S rRNA gene. J. Clin. Microbiol. 30: 796-800.
Grayston, 1. T., e.~C. Kuo, L. A. Campbell, and S.
Wang. 1989. Chlamydiapneumoniae sp. novo for Chlamydia sp. strain TWAR. Int. J. Syst. Bacteriol. 39: 88-90.
Higuchi, R. 1989. Simple and rapid preparation of samples for PCR. In: Erlich, H.A. (ed.) PCR Technology: Principles and applications for DNA amplification. pp. 31-38. Stockton Press, New York, London, Tokyo, Melbourne, Hongkong.
Holland, S.M., e.A. Gaydos and C. Quinn. 1990.
Detection and differentiation of Chlamydia trachomatis, Chlamydia psittaci and Chlamydia pneumoniae by DNA amplification. J. Infect. Dis. 162: 984-987.
Kaltenboeck, B., K.G. Kousoulas and J. Storz. 1991. Detection and strain differentiation of C. psittaci mediated by two-step polymerase chain reaction. Clin. Microbiol. 29: 1969-1975.
Kaltenboeck, B., K.G. Kousoulas and J. Storz. 1993. Structures of allelic diversity and relationships among the major outer membrane protein (ompA) genes of the four chlamydial species. J. Bacteriol. 175: 487-502.
Kaltenboeck, B. and 1. Storz. 1992. Biological properties and genetic analysis ofthe ompA locus in chlamydiae isolated from swine. Am. J. Vet. Res. 53: 1482-1487.
Mahony, J. B., K. E. Luinstra, 1. W. Sellors and M.A. Chernesky. 1993. Comparison of plasmidand chromosome- based polymerase chain reaction assay for detecting Chlamydia trachomatis nucleic acids. J. Clin. Microbiol. 31: 1753-1758.
Moulder, J. W., Hatch, T. P., Kuo, C.-C., Schachter, J. and Storz, J. 1984. Genus I. Chlamydia. Jones, Rake and Stearns 1945, 55AL. In : Krieg, N. R. and Holt, J. G. [eds.] Bergey's Manual of Systematic Bacteriology. voLl. pp. 729-739. Williams & Wilkins. Baltimore.
Pudjiatmoko, H. Fukushi, Y. Ochiai, Y. Yamaguchi and K. Hirai. 1996. Diversity of feline Chlamydia psittaci revealed by random amplification of polymorphic DNA. Vet. Microbiol. 54: 73-83.
Pudjiatmoko, H. Fukushi, Y. Ochiai, T. Yamaguchi and K. Hirai. 1997. Phylogenetic analysis of the genus Chlamydia based on 16S rRNA gene sequences. Int. J. Syst. Bacteriol. 47:425-431.
Schachter, J., H.B. Ostler and K.F. Meyer. 1969.
Human infection with the agent of feline pneumonitis. Lancet 1, 1063-1065.
Schachter, J. 1988. Chlamydiaceae: The Chlamydiae., p. 847-863. In : Lennette, E., P. Halonen, and F. A. Murphy (Eds.), Laboratory Diagnosis of Infectious Diseases. Principles and Practice, Vol. II: Viral, Rickettsial and Chlamydia I Diseases. Springer-Verlag, New York, Berlin, Heidelberg, London, Paris, Tokyo.
Stamm, W.E. 1988. Diagnosis of Chlamydia trachomatis genitourinary infections. Ann. Intern. Med. 108: 710-717.
Storz, J. and H. Krauss. 1985. Chlamydia. In: BIobel, H. and T. Schlieser (eds.). Handbuch der Bakteriellen Infektionen bei Tieren. pp. 446-531. VEB Gustav Fisher Verlag, Jena, Jena.
Thomas, R., H.C. Davison and AI. Wilsmore. 1990.
Use of the IDEL4 ELISA to detect Chlamydia psittaci (ovis) in material from aborted fetal membranes and milk from ewes affected by ovine enzootic abortion. Br. Vet. 1. 146: 364-367.
Valassina, M., M.G. Cusi, D. Corsaro, C. Buffi, G.
Piazzesi and V.P.E. 1995. Detection by multiplex polymerase chain reaction and typing of Chlamydia trachomatis isolates. FEMS Microbiol. Lett. 130: 205-2 10.





{kind=link}
